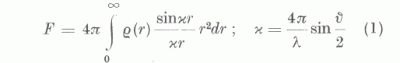
Fizikai Szemle honlap |
Tartalomjegyzék |
Röntgen és elektronsugarak szóródásának meghatározására szolgáló elméleti számítások egyik legfontosabb tényezője az atomalakfaktor.
Röviden össze akarjuk foglalni azokat a módszereket, melyekkel eddig az atomalakfaktorokat meghatározták és rá akarunk mutatni arra, hogy hogyan lehet az eddig legpontosabbnak ismert atomalakfaktorokat, melyeket a »self consistent field« módszer segítségével meghatározott sűrűségre támaszkodva számítottak ki, olyan elemekre általánosítani, melyekre hasonló számítások még nem ismeretesek.
Az atomalakfaktort a szóró atom elektroneloszlása határozza meg. Ha ρ-val jelöljük a szóró atom vagy ion (továbbiakban atom) elektronsűrűségét, akkor az atomalakfaktor, gömbszimmetrikus elektroneloszlást tételezve fel, a
képlettel határozható meg. Így az atomalakfaktor meghatározására lényegében annyi módszert ismerünk, ahány módszer az atomok elektroneloszlásának meghatározására ismeretes. λ a Röntgen-sugár hullámhossza és ν a szórási szög, r pedig az atommagtól mért távolság.
Atomok és ionok elektroneloszlásának meghatározását a legkönnyebben a statisztikus atomelmélet Thomas-Fermi módszerével végezhetjük el, mert az összes atomok és ionok elektroneloszlásának meghatározása két a rendszámtól és az ionizáció fokától független tabellázott függvényre vezethető vissza. Tekintve, hogy az atomalakfaktor értéke legjelentősebben az atom elektronjainak a maghoz közeleső részeken való eloszlásától függ és az atom belső részein a Thomas-Fermi módszerrel számított eloszlás igen jól átlagolja a valódi elektroneloszlást, így jó sikerrel használható különösen nehezebb atomok esetében az atomalakfaktor meghatározására.
Bragg és West, Debye és Bewilogua számoltak a Thomas-Fermi módszerrel számított elektroneloszlás alapján atomalakfaktorokat. A Thomas-Fermi elméletben használatos x = r/µ, változót bevezetve (µ = 0,8853 a0/Z1/3) és a
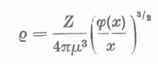
sűrűségeloszlást (1)-be helyettesítve nyerjük, hogy
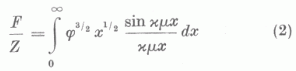
az u. n. redukált atomalakfaktor a
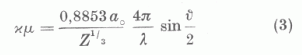
mennyiségnek a rendszámtól független függvényé. φ(x) a Thomas-Fermi egyenlet semleges atomokra ill. ionokra vonatkozó megoldása, a0 a legkisebb Bohr-féle elektronpályasugár a H-atomban. A módszer egyik igen nagy előnye, hogy az összes semleges atomok atomalakfaktorainak meghatározása egy univerzális függvény meghatározására vezethető vissza, mivel φ0(x), a Thomas-Fermi-egyenlet semleges atomokra vonatkozó megoldása a rendszámtól független, univerzális függvény. Így semleges atomra a számítások könnyen keresztülvihetők.
Ionokra és semleges atomokra vonatkozólag is Szőkefalvy-Nagy végzett számításokat, melyhez a Jensen által a statisztikus variációs módszerrel meghatározott elektroneloszlásokat használta fel. Pauling és Shermann szintén számítottak atomalakfaktorokat; ők az atom sűrűségeloszlását az egyes elektronok sajátfüggvényeinek segítségével építették fel, melyeket hidrogénszerűeknek választottak megfelelő árnyékolási állandókkal.
Az előzőknél lényegesen pontosabb elektroneloszlások azok, melyeket a »self consistent field« módszerrel számítottak. Így ezen számításokkal egyidőben merült fel annak szükségessége, hogy az atomalakfaktorok meghatározását is ezen elektroneloszlások alapján végezzék el. Többek között James és Brindley voltak azok, akik az ide vonatkozó számításokat elvégezték s számításaik szerint, bár a statisztikus elektroneloszlás alapján számítottól általában nem túl nagy eltérés adódott, mégis az eltérés egyes helyeken az 5%-ot is eléri s így feltétlenül tekintetbe kell venni.
»Self consistent field« számítások azonban csak aránylag kevés elemre ismeretesek s így James és Brindley is szükségesnek találták, hogy egy interpolációs eljárást dolgozzanak ki, mellyel oly atomok és ionok atomalakfaktorát is meghatározhassák, melyekre a sűrűségeloszlás nem ismeretes.
Az interpolációs eljárás lényege a következő: egy atom teljes elektronsűrűsége az egyes elektronok sűrűségeinek összege. Ezt tekintetbe véve (1) szerint az atom alakfaktora is az egyes elektronok alakfaktorának összegeként állítható elő. Az egyes elektronok alakfaktorai azonban próbálgatással olyan koordinátarendszerbe transzformálhatók, ahol azok univerzális lefutást mutatnak s így a transzformáló paramétert interpolálva megkapható a kiválasztott elektron hozzájárulása a teljes atomalakfaktorhoz. Az eljárást minden elektronnal végrehajtva a teljes atomalakfaktor meghatározható. A módszernek egy igen lényeges hiányossága azonban az, hogy az interpoláció kellő biztonsággal csak akkor hajtható végre, ha olyan rendszámokat vizsgálunk, melyek környezetében több »self consistent field« módszerrel végzett számítás már ismeretes. Lényegesen korlátozza az interpoláció alkalmazhatóságát az a tény, hogy kevés olyan elemre végeztek a »self consistent field« módszerrel számításokat, melyeknél a nagyobb főkvantumszámú állapotok is be vannak töltve úgy, hogy ezeknek interpolációja bizonytalan.
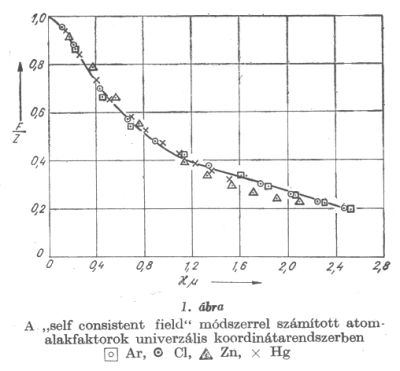
Ezek után nyilvánvaló annak a szükségessége, hogy a »self consistent field« módszerrel számított atomalakfaktorok általánosítására, azok más rendszámú elemekre való transzformációjára, konzekvens eljárást dolgozzunk ki.
Legyen egy semleges Z rendszámú atom potenciálja

ahol Zpe az atom effektív magtöltése természetesen még az r helykoordináta függvénye; e az elemi töltés. A Thomas-Fermi elmélet φ0(x) függvénye igen szoros összefüggésben van Zp-vel ugyanis, mint könnyen kimutatható
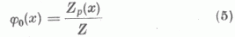
az u. n. redukált effektív magtöltést írja le, ha a helykoordinátát a Thomas-Fermi elmélet atomról-atomra változó µ = 0,8853 ao/Z1/3 egységében mérjük. Mint a statisztikus atomelméletből ismeretes φo/(x) az összes elemekre ugyanaz, tehát a rendszámtól független. Mint egy előző dolgozatában a szerző megmutatta Zp/Z a hullámmechanikai »self consistent field« módszer kicserélődés nélküli közelítésében is univerzálisnak tekinthető, ha az x koordinátát vezetjük be.
Felhasználva a gömbszimmetrikus sűrűségeloszlásra vonatkozó

Poisson-egyenletet, parciális integrálások után a redukált atomalakfaktorra nyerjük
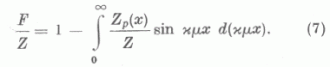
A (7) alak nyilvánvalóvá teszi, hogy a »self consistent field« módszerrel meghatározott atomalakfaktorok is jó közelítéssel xµ, univerzális függvényének tekinthetők. Az 1. ábrán néhány elem redukált atomalakfaktorát mutatjuk be ebben az univerzális koordinátarendszerben. xµ kifejezését felhasználva az ismeretes atomalakfaktorokból transzformációval azon elemekre is nyerhetünk atomalakfaktorokat, melyekre a »self consistent field« módszerrel még nem végeztek számításokat. A transzformáció természetesen annál jobb eredményre vezet, minél kisebb a transzformációban szereplő elemek rendszámai között a különbség. Szembetűnő az ábrán, hogy az atomalakfaktorok oly egymástól távol fekvő elemek esetében, mint a Hg és Ar is nagymértékben megegyeznek. A Zn atomnál a külső részeken tapasztalható aránylag nagyobb eltérés az Ar és Cl atomok atomalakfaktoraitól az elektronszerkezetbeli különbségre vezethető vissza. Az előbbi lezárt d héjjal, míg az utóbbiak lezárt ill. egy elektron hiánnyal rendelkező (s, p) héjjal rendelkeznek. A Zn atom alakfaktorához hasonló viselkedés nyomai a Hg-nál is kimutathatók. A transzformációnál az itt említett sajátságok is .figyelembe vehetők. Végül említsük meg, hogy az 1. ábrán látható függvények analitikus alakkal is közelíthetők s így pl. az (s, p) lezárt héjjal vagy ehhez hasonló elektronszerkezettel rendelkező atomok atomalakfaktorai számára a
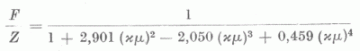
formula adható meg, mely a számítottól maximálisan 2%-al tér el.
A transzformáció ionok atomalakfaktorának meghatározására is felhasználható, itt csupán egy kissé bonyolultabb feladat előtt állunk, mert a transzformáció nem az azonos töltésű ionok között hajtható végre, hanem az azonos ionizációsfokú ionok-tehát melyekre a q = (Z -N)/Z mennyiség egyezik meg - atomalakfaktorai hozhatók egymással kapcsolatba.
Gáspár Rezső
Műszaki Egyetem Fizikai Intézete, Budapest