
Fizikai Szemle honlap |
Tartalomjegyzék |
Márk Géza, Vancsó Péter, Biró László Péter
MTA TTK Műszaki Fizikai és Anyagtudományi Kutatóintézet
A grafén a grafit egyetlen kristálysíkja, szénatomok hatszöges rácsba rendezett hálózata. Ez a 2004-ben előállított anyag, különleges tulajdonságai miatt, esélyes lehet a szilícium "leváltására" a nanoelektronikában. Ha grafént ipari skálán akarunk előállítani, erre a kémiai gőzfázisú leválasztás módszere (CVD) használható. Viszont ez a módszer "tökéletlen" grafént hoz létre, ugyanis a grafénsíkok sok kis kétdimenziós krisztallitból, doménből, állanak. Ebben a cikkben azt a kérdést járjuk körül, hogy ez a polikristályos grafén alkalmas-e nanoelektronikai célokra.
A grafitceruzát mindannyian ismerjük, tudjuk, ha a grafitot végighúzzuk a papíron, nyomot hagy. Ennek oka a grafit anyagszerkezetében rejlik: ez egy úgynevezett réteges anyag, ahol a rétegeken belül az atomok erősen kötődnek egymáshoz, ám a rétegek között gyenge van der Waals-kötés van, ezért a rétegek könnyen elválaszthatók egymástól. Le lehet-e választani egyetlen réteget a grafitból? Andre Geim és Konstantin Novoselov 2004-ben megmutatta, hogy ez valóban lehetséges, az így kapott egyetlen atomiréteg-vastagságnyi grafit neve grafén. Geim és Novoselov nemcsak létrehozták a grafént, hanem ennél sokkal többet tettek: okosan tervezett kísérletekkel megvizsgálták e leheletnél vékonyabb új anyag jellemzőit, és igen izgalmas, ígéretes tulajdonságokat találtak. Nem csoda, hogy ez a két kutató már 2010-ben elnyerte a fizikai Nobel-díjat [1].
G. E. Moore még 1965-ben észrevette [2], hogy a szilícium alapú integrált áramkörök "sűrűsége" (azaz az egységnyi felületre jutó áramköri elemek száma) exponenciálisan növekszik. Bizonyára Moore maga sem hitte volna, hogy ez a tendencia évtizedeken keresztül folytatódni fog [3]. Például egy mai kommersz flash memória eszköz ("pendrájv") 64 Gigabyte-os, ami azt jelenti, hogy néhány négyzetmilliméter területen 64 · 10243 · 8 = 5,5 · 1011 bitet tárol. De a szakértők szerint (www.itrs.net) az exponenciális növekedés nem tarthat akármeddig – a szilíciumtechnológia fizikai tulajdonságai 2020 körül megállítják a növekedést. Ezért a mikroelektronikai ipar – amely lassan nanoelektronikába megy át, hiszen egy mai integrált áramkör vonalszélessége 30 nanométer körül jár – keresi, mivel lehet majd a szilíciumot felváltani.
Egyik, talán legígéretesebb "jelölt" a grafén [4]. Nem csoda, hiszen a grafénben a töltéshordozók mozgékonysága 200 000 cm2/Vs körüli – több, mint százszor nagyobb, mint a szilíciumé. A grafén hővezető képessége is kiváló, ami megoldhatja azt a súlyos problémát, hogy a miniatürizálással együtt növekszik az integrált áramkörök által termelt hő.
Még nem beszéltünk arról, tulajdonképpen hogyan állította elő Geim és Novoselov az egyetlen atomi réteg vastag szénréteget? E módszer nagyszerűsége a hihetetlen egyszerűségében rejlik: ragasztószalagot nyomtak a grafitkristály felületéhez és az ezen fennragadt néhány atomréteg vastag graféndarabokat vékonyították tovább úgy, hogy ismételték a ragasztószalagos leválasztási trükköt, egészen addig, amíg egyetlen réteg maradt. Ezt a trükköt nem túl nehéz utánozni, a mi laboratóriumunkban, az MTA TTK Műszaki Fizikai és Anyagtudományi Kutatóintézetében is megcsinálta a 2010-ben "Junior Príma" díjjal kitüntetett Nemes-Incze Péter kollégánk, ahogy ez az 1. ábrán látszik.1
Ez a "tépéses" grafénelőállítási módszer kiválóan alkalmas laboratóriumi célokra, például graféntranzisztort is létrehoztak már így előállított grafénből. Ám ha a grafént ipari skálán (tonnaszámra) szeretnénk előállítani, akkor más módszer után kell néznünk! A manapság legígéretesebb ilyen módszer az úgynevezett kémiai gőzfázisú leválasztás (Chemical Vapor Deposition, CVD). A CVD módszerben valamilyen széntartalmú kép a CVD grafén doménszerkezetét mutatja. A domének határvonalai a képen feketék. A fehér vonalak a grafén felgyűrődései. Az egyes szemcsék kristálytani orientációját különböző – áttetsző – szürkeárnyalatok jelzik. (Nemes-Incze Péter (MTA TTK MFA) felvétele.) A különböző szürkeárnyalatok a különböző kristálytani orientációknak felelnek meg. (Philippe Lambin (Namuri Egyetem, Belgium) szimulációja.) gázt (például metánt) engednek egy magas hőmérsékletre (1000 °C) fűtött átmeneti fém (például réz) felületre. A folyamat paramétereinek (hőmérséklet, nyomás stb.) ügyes megválasztásával elérhető, hogy egyetlen grafitréteg (azaz grafén) keletkezzen a hordozó felületen.
A CVD módszerrel akár méteres grafénrétegeket lehet létrehozni, sőt a módszer folyamatos üzemre is alkalmas, amikor egy elméletileg "végtelen hosszú" és akár méternyi széles grafénszalagot hoznak létre. A CVD módszer tehát alkalmas arra, hogy olcsón és ipari méretekben állítsunk elő grafént. Ám van egy szépséghibája: a CVD módszerrel készült grafén polikristályos, azaz apró, 100-1000 nm méretű, szabálytalan alakú lemezkékből áll, mint a 2. ábra mutatja. A CVD grafén azért polikristályos, mert amikor a metán érintkezésbe kerül a rézfelülettel, egyszerre sok helyen indul meg a kristályosodás. Az így képződő kis grafénlemezkék mindaddig növekednek, amíg össze nem érnek (3. ábra), ekkor az érintkezési vonalon szemcsehatár alakul ki. A grafén szemcsehatár ugyanúgy krisztallitok közötti határ, mint a szilárdtestfizikában megszokott szemcsehatárok, de mivel a grafén kétdimenziós (2D) kristály, a szemcsehatár egydimenziós (1D), vonalszerű objektum lesz. A két, egymás felé növekvő, majd érintkező grafénszemcse kristálytani orientációja általában eltérő, a szemcsehatár mentén úgy kell elhelyezkednie a szénatomoknak, hogy a kétféle, eltérő orientációjú rács csatlakozni tudjon egymáshoz.
Természetesen a HOPG grafit tépésével kapott grafén rétegek sem tökéletes egykristályok, azok is tartalmazhatnak szemcsehatárokat. Csakhogy ebben az esetben a szemcsék egyrészt sokkal nagyobbak, másrészt a szemcsehatárok szabályosabbak. A cikk következő részében részletesen megvizsgáljuk, mit jelent ez a "szabályosság" és melyek a következményei.

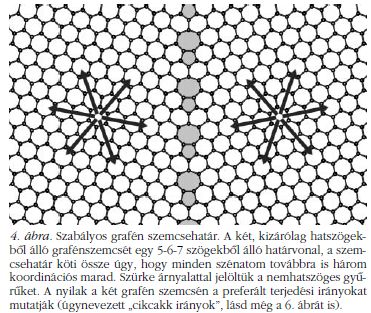
A tökéletes grafén egykristályban minden szénatomnak pontosan három szomszédja van és a szénatomok három kötése a grafén síkjába esik, a kötések egymással 120 fokos szöget zárnak be. A szénnek ezt a módosulatát nevezi a szaknyelv – bár talán kicsit pongyolán – "sp2 hibridizáció"-nak. A grafénben a szénatomok rácsa tökéletesen szabályos hatszögrácsot alkot. Mi történik, mikor két, egymás felé növekvő grafénszemcse összeér? Lehetséges-e úgy "összevarrni" a két rácsot, hogy "ne törjön meg az sp2 rács", azaz mindegyik szénatomnak továbbra is pontosan három szomszédja legyen? Igen, lehetséges, ehhez azonban feltétlenül szükséges, hogy a szemcsehatár mentén a rácsba ötszögeket és hétszögeket (sőt, esetleg négy-, nyolcszögeket!) építsünk be, hiszen ezáltal tud a grafénrács kristálytani iránya "elgörbülni". A 4. ábrán látunk egy ilyen szemcsehatár-konstrukciót. Az ilyen grafén szemcsehatárokat, ahol megmarad az sp2 rács, a továbbiakban "szabályos" szemcsehatárnak nevezzük.

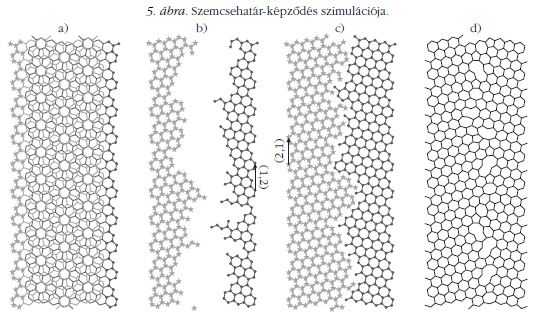
Ha azonban mélyebben szeretnénk megérteni, mi történik, amikor két növekvő grafénszemcse összeér és a valósághoz közelebb álló szemcsehatár-szerkezeteket szeretnénk létrehozni, akkor számítógépes szimulációt kell végeznünk. Belga kollégáinkkal együttműködve írtunk erre a feladatra egy Monte-Carlomódszeren alapuló programot. A program működése az 5. ábrán látható.
Azonban az így létrejött szimulált szemcsehatár-szerkezetekben általában megtörik az sp2 rács, azaz a szemcsehatárban nem csak hármas, hanem kettes koordinációjú szénatomok is előfordulnak. Sőt, a kétdimenziós rácsban helyenként kis "folytonossági hiányok", azaz vakanciák is keletkeznek, ráadásul nem csak 5-6-7, hanem 4-8 tagú széngyűrűk is előfordulnak. Az ilyen grafén szemcsehatárokat "szabálytalan" szemcsehatároknak hívjuk.
Napjainkban már ott tart a nanotudomány, hogy képesek vagyunk atomi felbontással vizsgálni az anyagok felületét. Ugyan ez csak bizonyos anyagokon és különleges körülmények között lehetséges, de mégis léteznek már atomi felbontású technikáink – például a pásztázó alagút-mikroszkópia (Scanning Tunneling Microscopy, STM) és a nagyfelbontású transzmissziós elektronmikroszkópia (High Resolution Transmission Electron Microscopy, HRTEM). A pásztázó alagútmikroszkóp elvi működése igen könnyen megérthető [5]: kell egy nagyon hegyes tű – ideális esetben csak egyetlen atom legyen a hegyén! – és egy olyan finom mozgató szerkezet, ami a tű hegyét nanométer alatti pontossággal képes mozgatni a vizsgált minta felülete fölött. A tű és a minta közé feszültséget kapcsolunk (volt nagyságrendben) és olyan közel visszük a tűt a felülethez, hogy még éppen ne érjen hozzá. Ha már elég közel értek egymáshoz, akkor megindul közöttük a kvantummechanikai alagútáram. Ezután a tűvel végigpásztázzuk a minta felületét és egy számítógép képernyőjén kialakul a minta atomi felbontású képe. A valóságban ahhoz, hogy ez működjön, természetesen sok gyakorlati nehézséget le kell küzdeni, például nagyon fontos a rezgéscsillapítás, a termikus drift kiküszöbölése, és így tovább.
Az STM és HRTEM mérések bebizonyították, hogy a HOPG grafit tépésével kapott grafénmintákban a szemcsehatárok többnyire "szabályos" szerkezetűek, de a CVD grafénmintákban legtöbbször "szabálytalanok".
Miért fontos, hogy milyen egy grafén szemcsehatár? Azért nagyon fontos, mert a szemcsehatárok lényegesen befolyásolják a grafén tulajdonságait – nemkülönben, mint a háromdimenziós kristályok esetén, ahol az anyag tulajdonságait szintén meghatározza a szemcsehatárok milyensége. A mérések szerint a CVD grafénben a töltéshordozók mozgékonysága több nagyságrenddel rosszabb, mint az egykristályos grafénben és ez rossz hír a nanoelektronikai alkalmazások szempontjából.
Ezért osztályunkon részletesen megvizsgáltuk, hogyan befolyásolja a szemcsehatárok atomi szerkezete azok elektronszerkezetét és transzporttulajdonságait. Megkerestük, hogy a szemcsehatár-szerkezet miféle tulajdonságai, jellemzői gyakorolnak hatást és pontosan milyen hatást a transzporttulajdonságokra.
Legfontosabb kísérleti eszközünk az alagútmikroszkóp (STM), annak topográfiai és spektroszkópiai módjában. Anélkül, hogy a részletekbe mennénk, csak anynyit fontos tudnunk, hogy az STM mind a minta felületének geometriájáról, mind elektronszerkezetéről atomi (vagy még finomabb) skálán szolgáltat információt. Ám a nanoskálájú mérések helyes értelmezéséhez elengedhetetlenül szükségünk van a számítógépes szimulációra, ugyanis a nanovilágról tudósító minden mérés közvetett mérés. A grafén szemcsehatárok elméleti vizsgálatában számos módszert használunk:
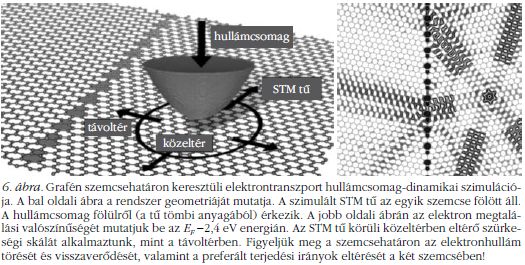
Azt már régóta tudjuk (Wallace számolta ki 1947-ben2), hogy a grafénsíkban az elektronok mozgása irányfüggő (anizotróp). Az anizotrópia annál kifejezettebb, minél inkább eltér az elektron energiája a Fermi-energiától.3 Ez azt jelenti, ha a grafén vezetőképességét úgy mérjük, hogy az egyik elektróda rögzített, a másik elektródával pedig egy körvonal mentén körbejárjuk azt, akkor a vezetőképesség szögfüggő. Az effektus gyakorlati kimutatásához alacsony hőmérsékleten és elég kicsi (mikronnyi méretű) mérőkörsugáron kell mérni. Mi történik, ha egy szemcsehatár két oldalára elektródákat helyezünk és mérjük a szemcsehatáron keresztüli vezetőképességet? A szemcsehatár mentén két, eltérő kristálytani orientációjú graféndarab találkozik. Az elektronok az egyik szemcséről a másikra akkor tudnak könnyen áthaladni, ha a preferált terjedési irányok a két szemcsében jó közelítéssel megegyeznek. Ha nem egyeznek meg a preferált terjedési irányok, akkor törési, visszaverődési jelenségek lépnek föl – ezek csökkentik az elektron átjutásának valószínűségét. Ilyen törést és visszaverést mutatunk be a 6. ábrán, hullámcsomag-dinamikai szimuláció [7] segítségével. Tehát megfogalmazhatjuk a felismerést: a grafén szemcsehatár vezetőképessége függ a két csatlakozó szemcse kristálytani orientációjának eltérési szögétől.
A szemcsehatárok elektronszerkezetének bemutatásához be kell vezetnünk az állapotsűrűség (Density of States, DOS) fogalmát. Ez a kondenzált anyagok fizikájában nagyon gyakran használt függvény azt mondja meg, hogy adott E energia körüli kis ΔE energia-intervallumban hány energiaállapot helyezkedik el. A DOS függvény ismeretében az adott anyag számos tulajdonságát ki tudjuk számítani, "meg tudjuk jósolni", például az elektromos és hővezetőképességét, optikai tulajdonságait stb. Egy adott anyag állapotsűrűsége két dologtól függ: az anyagot felépítő atomok fajtájától és elrendezésük mikéntjétől. (Szabályos kristályok esetén az atomi elrendezés térben periodikus, ezért itt az atomi elrendezést kristályszerkezetnek hívjuk.) A DOS függvényt kvantummechanikai módszerekkel (például DFT technikával) ki lehet számítani és számos módon meg is lehet mérni (például az STM műszer segítségével úgy, hogy változtatjuk a tűre adott feszültséget – ezt a technikát hívjuk alagút-spektroszkópiának).
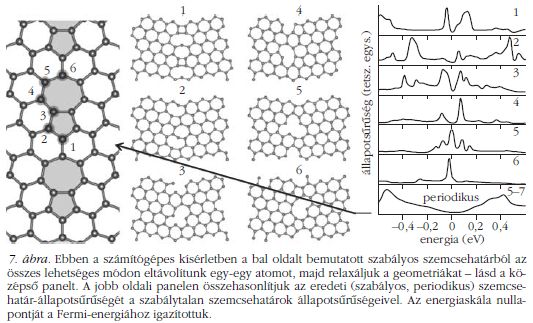
A 7. ábrán egy számítógépes kísérletet mutatunk be, amelynek segítségével megvizsgáltuk [8], hogyan függ a szemcsehatárok elektronszerkezete a geometriai szerkezetüktől. A kiinduló rendszer egy szabályos szemcsehatár, e szemcsehatár DOS függvényét láthatjuk a jobb alsó panelen. Ez egy "unalmas", sima függvény és a Fermi-energián nem tartalmaz állapotokat. Ahogy fentebb írtuk, a CVD grafénben általában szabálytalan szemcsehatárok fordulnak elő – olyanok, ahol sérül az sp2 rács, a szénatomok egy részének nincs három kötése, csak kettő. Ezt a helyzetet úgy modelleztük, hogy a szabályos szemcsehatárból kivettünk egy atomot, majd úgynevezett "geometriai relaxációt" végeztünk. A geometriai relaxáció azt jelenti, hogy az egyes atomokra ható erőket figyelembe véve hagyjuk az atomokat elmozdulni mindaddig, amíg az összes atom meg nem találja egyensúlyi helyzetét. Így voltaképpen a rendszer mechanikai energiáját minimalizáljuk,4 amire azért van szükség, mert egy atom mesterséges eltávolításával felborul az egyes atomokra ható erők egyensúlya, tehát meg kell keresnünk az új egyensúlyi konfigurációt. Erre a relaxált geometriára azután ismét kiszámoljuk az elektronok állapotsűrűségét. Egy egyszerű végtelen grafénsíkban az összes szénatom környezete pontosan azonos – egyforma az összes kötésszög (120 fok) és kötéstávolság (0,142 nm). Ám egy szemcsehatárban több, különböző helyzetű atom található, amelyeknek "szomszédság szerkezete" (kötéseik száma, szöge és távolsága) eltér egymástól. Azért, hogy teljes képet kapjunk a rendszer viselkedéséről, az összes lehetséges módon eltávolítottunk egy-egy szénatomot, mindegyik esetre elvégeztük a geometriai relaxációt és az állapotsűrűség-számítást. Az így kapott szemcsehatár-geometriákat és DOS függvényeket mutatja be a 7. ábra.
A szabálytalan szemcsehatárokra kapott DOS függvények bonyolult szerkezetűek és jócskán különböznek egymástól. Mégis, mi bennük a közös? Egyrészt mindegyik szabálytalan szemcsehatárra számolt DOS függvény sok csúcsot tartalmaz, szemben a szabályos szemcsehatárra kapott sima függvénnyel. Másrészt megfigyelhetjük, hogy a szabálytalan szemcsehatárok jelentős értékű állapotsűrűséggel rendelkeznek a Fermi-energia körüli energiatartományban, holott a szabályos szemcsehatár állapotsűrűsége itt igen kicsi. Vannak olyan atomi elrendezések is, ahol magán a Fermi-energián is csúcsot látunk!
Amikor az elektron áthalad egy szabálytalan grafén szemcsehatáron, ezekkel a nagy állapotsűrűségű, a Fermi-energiához közeli elektronállapotokkal találja magát szembe, ezeken kell "átküzdenie magát", ami ritkán sikerül neki. Tehát kicsi az átmeneti, viszont nagy a visszaverődési valószínűség, ezért mondjuk, hogy ezek a szemcsehatárra lokalizált állapotok úgynevezett szórócentrumok. Végeredményben drasztikusan lecsökken a szemcsehatár vezetőképessége. Ezáltal sikerült megértenünk azt a kísérleti tapasztalatot, hogy a CVD grafén vezetőképessége messze elmarad a "tépett" grafén vezetőképességétől.
A címben feltett kérdésre "feltételes igennel" tudunk válaszolni. Megmutattuk, hogy a grafén szemcsehatárok elektromos tulajdonságai erősen függenek a szerkezetüktől. Azok a szemcsehatárok, ahol megmarad a szénatomok hármaskötésű hálózata (sp2 rács), sokkal kisebb akadályt jelentenek az elektronok számára, mint azok a szemcsehatárok, ahol sérül a hármaskötésű hálózat, azaz kettős koordinációjú szénatomok és vakanciák jelennek meg – ugyanis ezek erős szórócentrumot képeznek az elektronok terjedése számára. Ahhoz, hogy a kémiai gőzfázisú leválasztással (CVD módszer) előállított grafénben óhatatlanul létrejövő szemcsehatárok ne rontsák le drasztikus módon a grafén elektromos tulajdonságait, szükséges volna a CVD technológia paramétereinek jobb kézbentartása, finomhangolása ("szemcsehatár mérnökség"), ami által olyan szemcsehatárokat lehetne létrehozni, amelyek "barátságosabban" viselkednek az elektromos transzport szempontjából, azaz kevesebb szórócentrumot tartalmaznak.
___________________
A 2013. évi Magyar Fizikus Vándorgyűlésen elhangzott előadás írott változata. További információk: http://www.nanotechnology.hu___________________