Fizikai Szemle honlap |
Radnai Gyula
ELTE, Általános Fizika Tanszék
pV = RT (n = 1 mólra)
Az ideális gáz belső energiáját pedig az alábbi kalorikus állapotfüggvény szolgáltatja:U = U (p, V)
A fenomenologikus termodinamikának főtételekre alapozott tárgyalásakor a nulladik főtétel biztosítja a hőmérséklet egzisztenciáját, az első főtétel pedig a belső energia, egzisztenciáját minden termodinamikai rendszerre. Így még a második főtétel előtt definiálhatunk egy empirikus hőmérsékleti skálát, valamelyik termodinamikai rendszer termikus állapotegyenletének felhasználásával. A szokásos eljárás az, hogy referencia rendszernek éppen az ideális gázt választjuk, mert a pV = RT állapotegyenletben a gázok anyagi minőségétől való függés nem szerepel. (Joggal választhatnánk például az ideális paramágneses szilárd anyagot is; az ideális gáz választásának történeti oka van.) A második főtétel az entrópia egzisztenciáját biztosítja. Kiterjesztve az entrópia fogalmát összetett rendszerek nem egyensúlyi állapotaira is, a második főtétel segítségével a folyamatok irányára is következtethetünk. Csupán egyensúlyi állapotokon keresztül történő idealizált folyamatok esetén is lehetővé válik azonban a második főtétel alapján egy olyan univerzális hőmérsékleti skála definiálása, amely független a hőmérsékletet mérő anyag állapotegyenletétől:![]()
pV = RT
úgy, hogy T most a második főtétel segítségével definiált abszolút hőmérséklet.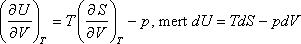
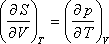
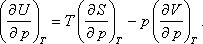
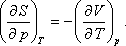
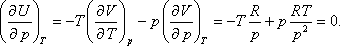
pV=RT
egyenlet nem más, mint T definíciója, vagyis a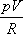
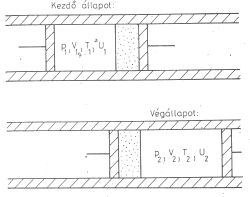
p1; V1 = V; T1; U1
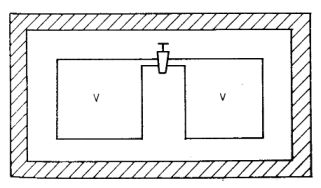
1. ábra
p2; V2 = 2V; T2; U2,
Állítás: U1 = U2U2 - U1 = Q + W
Most Q = 0 az adiabatikus szigetelés miatt, W = 0 mert a termodinamikai munka külso munka, s a környezet nem végzett a gázon munkát. Akármilyen gázzal is végezzük el a Gay-Lussac kísérletet, a gáz új egyensúlyi állapota tehát mindig ugyanolyan belső energiájú, mint a kiindulási állapot volt. A hőmérsékletet mérni kell. Gay-Lussac úgy végezte el a mérést, hogy az edényeket folyadékfürdőbe állította, s azt mondta, hogy ha T2 ≠ T1, akkor ez megváltoztatja a folyadékfürdő hőmérsékletét. (Elvileg igen, a hőkapacitások közti óriási különbség miatt azonban a gáz hőmérsékletváltozása gyakorlatilag mérhetetlenül kis változást okoz a folyadékfürdő hőmérsékletében). Gay-Lussac nem kapott hőmérsékletváltozást, ezért arra következtetett:T2 = T1
Ma már tudjuk, hogy az állítás helyes, de mint a fenti kísérletből levont mérési következtetés nem fogadható el. Gay-Lussac kísérletének mégis fizikatörténeti jelentősége van. Az a felismerés, hogy az ideális gáz szabad tágulásakor hőmérséklete nem változik meg, mind az energiafogalom kialakulásában, mind az ideális gáz kinetikus modelljének felállításában fontos szerephez jutott. Idézzük csak fel Robert Mayer nevezetes gondolatkísérletét! A hő és a munka közti kapcsolat felismeréséhez vezető egyik fontos gondolata az volt hogy amennyivel több hő kell az ideális gáz 1°-kal való felmelegítéséhez állandó nyomáson mint állandó térfogaton, annyi munkát végez a gáz az izobár tágulás közben. Dehát ebben a kétféle folyamatban azonos kezdőállapot esetén a gáz két különböző végállapotba jut! A következtetés pedig csak akkor jogos, ha ugyanabból a kezdőpontból ugyanabba a végállapotba jut mindkét esetben a gáz. Ehhez a kiegészítéshez volt szüksége Robert Mayernek Gay-Lussac kísérletére (2. ábra). Az 1 állapotból kétféle módon visszük át a gázt a 2 állapotba:Qp - QV, = CpΔT - CVΔT
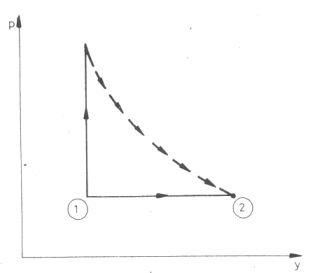
2. ábra
W = pΔV = RΔT mert a folyamat izobár.
Így jutunk az azóta már Robert Mayerről elnevezett összefüggéshez:Cp - CV = R.
Semmi kétség: a Gay-Lussac kísérlet irreverzibilis folyamat, (ezért is ábrázoltuk a folyamatot a pV diagramon szaggatott vonallal). A kezdő és végállapot azonban egyensúlyi, ezért kiszámítható az entrópianövekedés. Ez ugyanannyi, mintha például hőtartályba ágyazva, izotermikusan vinnénk az ideális gázt a megadott kezdőállapotból a megadott végállapotba, tehát
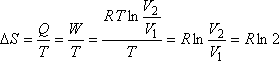 |
||
| (ha V2 = 2V1) |
{hőtartály + gáz}
összetett rendszer entrópiája nem változik, ahogyan azt egyensúlyi folyamatok során el is várjuk. Ebben az egyensúlyi folyamatban a gáz és a hőtartály entrópiaváltozása "kompenzálja egymást" (Clausius megfogalmazása), míg az irreverzibilis szabad tágulás során a gáz entrópianövekedését nem kompenzálja semmi a környezetben. A gáz entrópiája mindkét esetben ugyanannyit változik, mivel a gáz ugyanabból a kezdőállapotból ugyanabba a végállapotba jut, s az entrópia - állapotjelző. Fizikatörténeti háttér A Gay-Lussac-kísérlettel nem az a baj, hogy irreverzibilis, hanem az, hogy a mérési pontosság nem volt megfelelő. Ezen igyekezett javítani Joule. Robert Mayer 1845-ben adta ki német nyelven azt a munkáját, amelyben fent ismertetett gondolatmenetét publikálta s amelyben Gay-Lussac 1807-es kísérletére támaszkodott. Joule 1843-ban végezte el Gay-Lussac kísérletének módosított változatát a következő módon. A két azonos térfogatú edényt fémből készítette el, és külön-külön kaloriméterekbe helyezte el őket. A kaloriméterek térfogata csak kevéssel volt nagyobb, mint a fémedényeké, így viszonylag kis kapacitású volt a folyadék, ami a fémedényeket a kaloriméterekben körülvette. Ő is a folyadékok hőmérsékletváltozását mérte, s ebből következtetett a gáz hőmérsékletváltozására. A kiindulási állapotban az egyik edényt vákuumra szivattyúzta le, a másik edénybe pedig annyi levegőt pumpált, hogy nyomása 22 atmoszféra lett. Ezután kinyitotta a csapot, s a következő hőmérsékletváltozásokat tapasztalta: azon az oldalon, ahol eredetileg a 22 atmoszférás levegő volt, a hőmérséklet 2,36 F°-kal lett kisebb, a másik oldalon a hőmérséklet 2,38 F°-kal lett magasabb. [5] Mivel a két kaloriméter hőkapacitása egyenlő volt, ez azt jelenti, hogy ha a mechanikai egyensúly után sokkal lassabban beáll majd a termikus egyensúly is a két edényben levő gáz között, akkor a véghőmérséklet a mérési pontosságon belül meg kell hogy egyezzék a kiindulási hőmérséklettel. Joule körültekintő kísérletező ügyessége tette lehetővé, hogy a Gay-Lussac kísérlet visszanyerte eredeti bizonyító erejét. Már csak egyedül Joule nem volt megelégedve a mérési pontossággal. Az az igazság, hogy Joule, amikor eredeti formájában - tehát egyetlen közös kaloriméterben - ismételte meg Gay-Lussac kísérletét, mégiscsak kapott valamennyi hőmérsékletváltozást, de ez a mérési hiba határát súrolta. Azóta, hogy 1807-ben Gay-Lussac megfogalmazta nevezetes tételeit a gázok egyenlő viselkedéséről a kísérleti módszerek finomodásával egyre több kételkedő kutató lépett fel Gay-Lussac kijelentését cáfoló mérési adatokkal. A Francia Akadémia már 1811-ben pályázatot hirdetett a minél pontosabb kísérleti vizsgálatokra. Joule-ban két nézet viaskodott egymással. Egyik a gázok egyforma viselkedését értelmezni tudó molekuláris elmélet kidolgozásának reménye, amely szerint CV állandó. Másik a saját és mások kísérletei alapján felmerülő kétely CV állandóságában. Hogyan növelhető a fajhőmérések pontossága? Joule 1848-ban, Manchesterben egy tudományos felolvasóülésen kifejti azt a véleményét, hogy "azok a kutatások, melyeket Victor Regneault a francia kormány megbízásából vállalt a testek hőkapacitásának fontos problémáját is felölelik, így rövid időn belül új meghatározást várhatunk a gázok fajhőjére. Ezt a meghatározást ugyanaz a pontosság fogja jellemezni, amely teljesen megérdemelten szerzett dicsőséget ennek a híres kutatónak." [6] Ennél a híres kutatónál dolgozott néhány hónapig 1845-ben, miután Cambridge-ben matematikából és fizikából diplomázott - William Thomson. A Regnault által követelt nagy mérési pontosság indította Thomsont azokra a vizsgálatokra, amelyek az abszolút hőmérsékleti skála felállításához vezettek. Megszületett a tökéletes gáz absztrakciója, melyet a valódi gázok csak több-kevesebb pontossággal közelítenek. Itt Párizsban ásta ki a jótékony(?) feledés süllyesztőjéből a 21 éves Thomson az akkor már 13 éve halott Carnot-nak 1824-ben (Thomson születésének évében) publikált dolgozatát, és hazatérve Glasgowba 1848-ban közölte a termodinamikai hatásfokra alapozott abszolút hőmérsékleti skálát. Glasgow és Manchester mintegy 300 km-re vannak egymástól. Thomson és Joule szelleme számára ennél nagyobb távolság sem lett volna akadály barátságuk kialakulásához. Joule hat évvel volt idősebb Thomsonnál, először 1847-ben találkoztak [7]; lehet, hogy éppen Thomson hívta fel Joule figyelmét Regnault pontos méréseire. Ettől kezdve állandó levelezésben álltak egymással. Ha Thomson egyetemi előadásán valami új, frappáns demonstrációs kísérletet talált ki, azonnal megírta Joule-nak. Ugyanakkor Thomsont, akit lenyűgözött Fourier munkássága, amely még a hőanyag modelljével írta le matematikailag a hővezetés elméletét, egyedül Joule kísérleti eredményei győzték meg a hőanyagelmélet tarthatatlanságáról. [7, 8] Közös munkájuk legszebb eredménye annak az effektusnak a felfedezése, mellyel ipari méretekben megindulhatott a gázok cseppfolyósítása, s amely ma valamennyi háztartási hűtőgép működésének egyik nélkülözhetetlen feltétele, az un. Joule-Thomson effektus.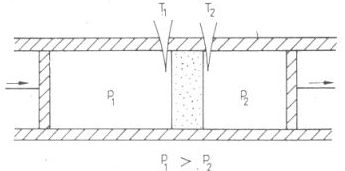
3. ábra
A henger két oldalán 1-1 adiabatikus dugattyút mozgatunk úgy, hogy a porózus fal két oldalán a nyomás állandóan p1 illetve p2 maradjon, miközben a porózus falon keresztül a gáz lassan áramlik a nagyobb nyomású oldalról a kisebb nyomású oldalra. (A porózus fal szerepe az, hogy ugyan megengedi a gáz átáramlását, de nem engedi a gázt felgyorsulni. Mintegy "lefojtja" a gáz áramlását, ezért a kísérletet gyakran "fojtásos" kísérletnek is nevezik. A fojtás szerepét egy szűkület is átveheti a csőben, a gyakorlati alkalmazás során gyakran így valósítják meg az effektust.) A porózus fal két oldalán termoelemekkel mérjük a T1 és T2 hőmérsékleteket. A kísérlet irreverzibilis, bár a gáz kezdő és végállapota (amikor csak a baloldalon illetve csak a jobboldalon helyezkedik el a kiszemelt gázmennyiség) egyensúlyi állapot. Ezt az alábbi ábrákkal lehet szemléltetni (4. ábra). Állítás: U1+ p1Vl = U2 + p2V2 Bizonyítás: Az első főtétel értelmébenU2 - U1= Q + W
Most Q = 0 az adiabatikus szigetelés miatt, W = p1V1 - p2V2 mert a baloldali dugattyú lenyomásakor p1V1 munkát végzünk, a jobboldali dugattyú elmozdulása közben pedig p2V2 munkát nyerünk. A befektetett munka így a kettő különbsége. Átrendezés után kapjuk a bizonyítandó állítást. A H = U + pV mennyiséget entalpiának nevezzük, így a Joule-Thomson kísérletbenH1 = H2
4. ábra
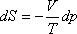
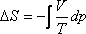
dp < 0.
kell, hogy legyen. Tekintettel arra, hogy V > 0, és T > 0, ezért szükségképpenΔS > 0.
Egy mól ideális gázra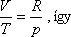
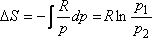
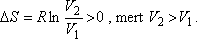
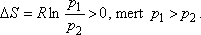
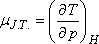
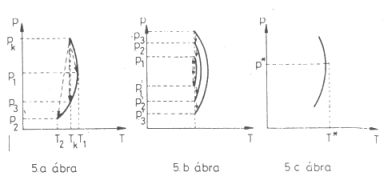
5. ábra. Állandó entalpiájú (izentalp) görbék és fojtásos folyamatok ábrázolása a (p; T) koordinátarendszerben |
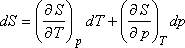
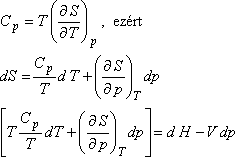
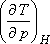 -t
akarjuk meghatározni:
-t
akarjuk meghatározni:
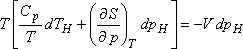
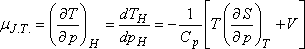
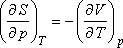
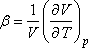
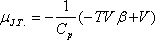
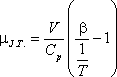
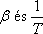 viszonyával tudjuk összekapcsolni.
Az anyagnak azokban az állapotaiban, ahol
viszonyával tudjuk összekapcsolni.
Az anyagnak azokban az állapotaiban, ahol
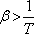 a Joule-Thomson-koefficiens pozitív, tehát a differenciális Joule-Thomson-effektus során az anyag
hőmérséklete csökken.
a Joule-Thomson-koefficiens pozitív, tehát a differenciális Joule-Thomson-effektus során az anyag
hőmérséklete csökken. 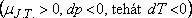 Ha viszont
Ha viszont
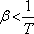 ,
akkor az anyag melegszik. Abban az esetben, amikor
,
akkor az anyag melegszik. Abban az esetben, amikor
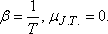 .
Ez éppen az ideális gáz esetén valósul meg, hiszen az ideális gázra
.
Ez éppen az ideális gáz esetén valósul meg, hiszen az ideális gázra
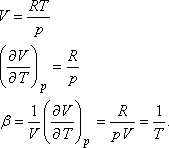
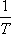 -hez
képest rendkívül kicsi (0 °C és 4 °C között negatív, 4 °C-on zérus, 4 °C és
100 °C között pozitív - 1 atm nyomáson), ezért jó közelítésben a JouleThomson koefficiens
vízre:
-hez
képest rendkívül kicsi (0 °C és 4 °C között negatív, 4 °C-on zérus, 4 °C és
100 °C között pozitív - 1 atm nyomáson), ezért jó közelítésben a JouleThomson koefficiens
vízre:
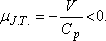
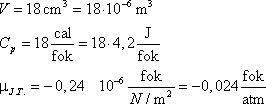
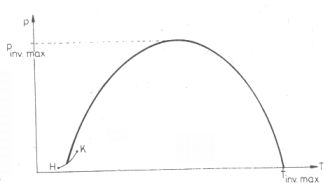
6. ábra. Inverziós görbe a (p; T) koordinátarendszerben.
H = Hármaspont (a gáz, a folyadék és a szilárd fázis közös egyensúlyi állapota),
K = Kritikus pont (a H-t K-val összekötő, ún. folyadék-gáz fázishatárgörbén
vannak a telített gőz állapotok)
pinv.max. = a legnagyobb inverziós nyomás
| Tkr | Tinv.max | Tinv.max/Tkr | |
| N2 | 126 K | 621 K | 4,9 |
| levegő | 132 K | 603 K | 4,6 |
| Ar | 151 K | 723 K | 4,8 |
| H2 | 33,3 K | 202 K | 6,1 |
| He | 5,2 K | 40 K | 7,7 |
| pkr | pinv.max | pinv.max/pkr | |
| N2 | 33,5 atm | 400 atm | 12 |
| H2 | 12,8 atm | 106 atm | 13 |
| He | 2,3 atm | 37 atm | 16 |
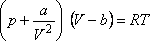
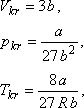
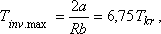
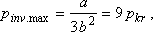
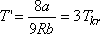
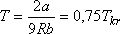 -nál. (Emlékezzünk vissza, hogy H2O esetén a mérési
adatok szerint az inverziós görbe 507 K-nél metszi a folyadék-gőz fázishatárgörbét. Ez a H2O kritikus
hőmérsékletének - 647 K-nek 0,78-szorosa).
Összehasonlítva a van der Waals állapotegyenlet által jósolt
-nál. (Emlékezzünk vissza, hogy H2O esetén a mérési
adatok szerint az inverziós görbe 507 K-nél metszi a folyadék-gőz fázishatárgörbét. Ez a H2O kritikus
hőmérsékletének - 647 K-nek 0,78-szorosa).
Összehasonlítva a van der Waals állapotegyenlet által jósolt
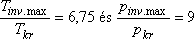
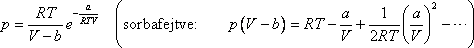
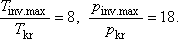
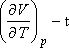 kell meghatároznunk, mivel
µJ.T. = 0,
ha
kell meghatároznunk, mivel
µJ.T. = 0,
ha 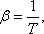 , vagyis
, vagyis
 A van der Waals állapotegyenlet:
A van der Waals állapotegyenlet:
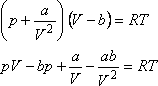
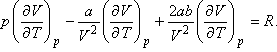
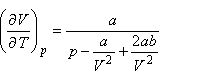
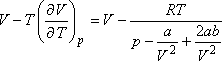
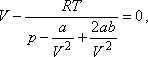
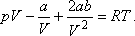
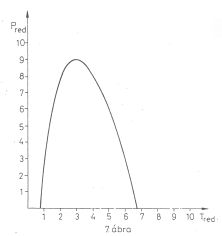
7. ábra A Van der Waals gáz inverziós görbéje;
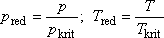
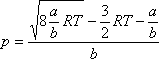
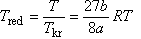
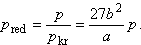
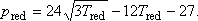
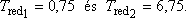
pred.max = 9
. A van der Waals inverziós görbe menete a 7. ábrán látható. Gyakorlati alkalmazások A múlt század utolsó évtizedeiben a gázok cseppfolyósításának nagyszabású programja többek között. éppen a Joule-Thomson-effektus felfedezése nyomán bontakozhatott ki. Ugyancsak a Joule-Thomson-effektust használják fel napjakban az ipari és háztartási hűtőgépekben, valamint a légkondicionáló berendezésekben. Mindezek tárgyalása azonban már meghaladná e cikk kereteit, a gyakorlati alkalmazások témája külön tanulmányt érdemel.