A JOULE-THOMSON EFFEKTUS
Radnai Gyula
ELTE, Általános Fizika Tanszék
1. Az ideális gáz belső energiája
Az állandó tömegű ideális gáz egyszerű termodinamikai rendszer, mivel egyensúlyi állapotát két független állapotjelző egyértelműen
meghatározza [1,2].
Ha független állapotjelzőkül a nyomást és a térfogatot választjuk, akkor az ideális gáz hőmérséklete az alábbi termikus
állapotegyenletből kapható meg:
pV = RT (n = 1 mólra)
Az ideális gáz belső energiáját pedig az alábbi kalorikus állapotfüggvény szolgáltatja:
U = U (p, V)
A fenomenologikus termodinamikának főtételekre alapozott tárgyalásakor a nulladik főtétel biztosítja a hőmérséklet egzisztenciáját, az első főtétel pedig a belső energia, egzisztenciáját minden termodinamikai rendszerre. Így még a második főtétel előtt definiálhatunk egy empirikus hőmérsékleti skálát, valamelyik termodinamikai rendszer termikus állapotegyenletének felhasználásával.
A szokásos eljárás az, hogy referencia rendszernek éppen az ideális gázt választjuk, mert a pV = RT állapotegyenletben a
gázok anyagi minőségétől való függés nem szerepel. (Joggal választhatnánk például az ideális paramágneses szilárd anyagot is; az
ideális gáz választásának történeti oka van.)
A második főtétel az entrópia egzisztenciáját biztosítja. Kiterjesztve az entrópia fogalmát összetett rendszerek nem egyensúlyi
állapotaira is, a második főtétel segítségével a folyamatok irányára is következtethetünk. Csupán egyensúlyi állapotokon keresztül
történő idealizált folyamatok esetén is lehetővé válik azonban a második főtétel alapján egy olyan univerzális hőmérsékleti skála
definiálása, amely független a hőmérsékletet mérő anyag állapotegyenletétől:

Vajon a pV = RT állapotegyenlettel definiált "ideális gáz hőmérsékleti
skála" alkalmas-e olyan univerzális, "abszolút"
hőmérsékleti skálának, amelynek létezését a második főtétel mondja csak ki? Igen.
Egy közvetett bizonyíték lehet a következő eljárás:
- Feltesszük, hogy ideális gázra
pV = RT
úgy, hogy T most a második főtétel segítségével definiált abszolút hőmérséklet.
- Bebizonyítjuk a második főtétel felhasználásával, hogy ebben az esetben az ideális gáz belső energiája független a gáz
térfogatától és nyomásától, csak abszolút hőmérsékletétől függhet. A térfogattól való függés:
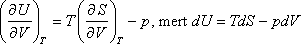
Felhasználva a következő Maxwell relációt:
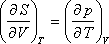
kapjuk:
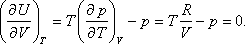
A nyomástól való függés:
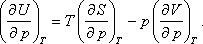
Felhasználhatjuk a következő Maxwell relációt:
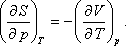
Így kapjuk:
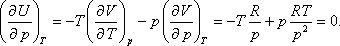
- Feltesszük, hogy ideális gázra a
pV=RT
egyenlet nem más, mint T definíciója, vagyis a
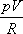
mennyiség egy empirikus, úgynevezett "ideális gáz hőmérséklet", és e definíció alapján gőzhőmérőt készíthetünk.
- Megvizsgáljuk kísérletileg, vajon igaz-e, hogy az ideális gáz belső energiája csak a pV szorzattól függ.
Vagyis megvizsgáljuk, hogy igaz-e a 2. pontban bebizonyított állítás akkor is, ha a pV = RT-ben T az ideális
gázzal hitelesített hőmérsékleti skála.
Egyik ilyen kísérlet az, amikor összehasonlítjuk az ideális gáz két tetszőleges olyan egyensúlyi állapotát, melyben belső energiája
(és mólszáma) egyenlő, de nyomása és térfogata is más-más értékű.
Megmérjük mindkét állapotban a hőmérsékletet (empirikus, ideális gáz skálán), és megvizsgáljuk, egyenlő-e a két hőmérséklet.
Ha igen, akkor ez azt jelenti, hogy az ideális gáz belső energiája csak a pV = RT-vel definiált empirikus, ideális gáz
hőmérséklettől függ.
Elvileg megfelelő kísérlet erre az, amelyet először (1807-ben) Gay-Lussac végzett el, majd Joule a múlt század 40-es éveiben
megismételt, részleteiben is megvizsgált.
- Ha kísérletileg is megfelelő igazolást nyer az a feltételezés, hogy az ideális gáz belső energiája csak az ideális gáz
hőmérséklettől függ, akkor ebből már az első főtétel alapján következik, hogy az ideális gáz egy izotermikus folyamatban
ugyanannyi hőt vesz fel, mint amennyi munkát végez, s egy Carnot körfolyamatban a felvett és a leadott hők abszolút értékeinek
hányadosa az ideális gáz hőmérsékletek hányadosával egyezik meg.
Ugyanez igaz azonban a második főtétellel definiált abszolút hőmérsékletek hányadosára is!
Ez azt jelenti, hogy az ideális gáz hőmérséklet arányos az abszolút hőmérséklettel.
Kényelmi okokból az arányossági tényezőt 1-nek választjuk, s így az ideális gáz hőmérsékleti skála egyben abszolút hőmérsékleti
skála lesz.
(Mechanikában hasonló eljárást követünk akkor, amikor a gravitációs és a tehetetlen tömeg arányosságát bizonyító kísérletek -
Eötvös, Renner, Dicke és mások kísérletei - közös eredményeként, egyenlőnek választjuk a kétféle módon definiált tömeg
mértékegységét és mértékszámát.)
Fizikatörténeti háttér
Gay-Lussac a gázok közös viselkedését kutatta, az ideális gáz fogalma akkor még nem létezett. éppen az ő vizsgálatai vezettek
azonban az ideális gáz absztrakciójához. Felállította alaptörvényeit, amelyeket 1807-ben saját és mások kísérleti tapasztalataira
támaszkodva a következőképpen mondott ki: [3].
- Valamennyi gáz eredeti térfogatának 1/267 részével terjed ki, ha hőmérsékletét 1 fokkal növeljük;
- A gázok kitágulása független a nyomástól.
Érdemes megemlíteni, hogy Gay-Lussac már öt évvel korábban, 1802-ben publikálta a gázok
hőtágulására végzett kísérleteinek eredményét. [4] Kísérleteit levegővel,
oxigénnel, nitrogénnel és hidrogénnel végezte Laplace felkérésére, aki erősen hitt a gázok egyforma
viselkedésében. Gay-Lussac 1799-től volt az Ecole Normale Politechnique hallgatója, s Laplace
1794 óta tanított ott. Az első, 1802-es publikációt követően Gay-Lussacot nevezetes, léggömbön
végzett kísérletei (1804, Biot-val közösen), majd olaszországi és németországi tudományos útjai
(1805, Alexander Humboldttal közösen) kötötték le. 1806-ban tért újra vissza Berlinből Párizsba,
ahol 1809-től lett az Ecole Normale Politechnique kémiatanára. Az 1807-es publikációt megelőző
években Dalton és Davy Angliában, Volta pedig Itáliában végzett hasonló méréseket.
A hőanyagelmélet teljes virágzását élte, forradalmian új gondolatok kémiában születtek ekkor. A
flogiszton-elméletet Lavoisier kvantitatív kémiai kísérletei döntötték meg, az anyag atomos
felépítésének hipotézise ekkor kezdett erőre kapni. Először a gázok kémiai reakcióinak magyarázatára
született meg Dalton atom és molekulahipotézise. Ebbe a kutatásba kapcsolódott be Gay-Lussac is,
ezért volt mindkettőjük számára fontos, hogy a gázok azonos tulajdonságait keressék.
A gázok egyforma hőtágulása mellett Gay-Lussac felállította a gázok egyforma fajhőjének hipotézisét
is. Pontosabban azt állította, hogy a gázok fajhője nem függ a gázok sűrűségétől (térfogatától). Ennek
igazolására végezte el azt a kísérletet, melyet a következőkben ismertetünk. Ez a kísérlet egyrészt a
második főtételt alátámasztó egyik közvetett bizonyítéknak tekinthető, másrészt az első főtétel
felfedezéséhez vezető egyik fontos lépés is volt a fizika történetében.
2. Gay-Lussac kísérlete - mai értelmezéssel
Vegyünk két azonos V térfogatú edényt, melyek a csappal vannak összekötve
(1. ábra).
Az egész rendszer legyen kívülről adiabatikusan szigetelve, ezt jelképezi az ábrán ferdén vonalkázott
keret. Kezdetben legyen ideális gáz csupán a baloldalon (a jobboldalon tehát vákuum van), így a
kezdőállapot jellemzői:
p1; V1 = V; T1;
U1
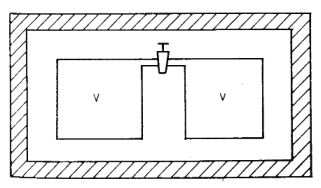
1. ábra
Nyissuk ki a csapot! Irreverzibilis, nem egyensúlyi állapotokon keresztül történő folyamat során a gáz kiterjed.
A folyamat végére az ideális gáz újra egyensúlyi állapotba kerül, e végállapot jellemzői:
p2; V2 = 2V; T2; U2,
Állítás: U1 = U2
Bizonyítás: Az első főtétel értelmében
U2 - U1 = Q + W
Most Q = 0 az adiabatikus szigetelés miatt, W = 0 mert a termodinamikai munka külso munka, s a környezet
nem végzett a gázon munkát.
Akármilyen gázzal is végezzük el a Gay-Lussac kísérletet, a gáz új egyensúlyi állapota tehát mindig ugyanolyan belső energiájú,
mint a kiindulási állapot volt. A hőmérsékletet mérni kell.
Gay-Lussac úgy végezte el a mérést, hogy az edényeket folyadékfürdőbe állította, s azt mondta, hogy ha
T2 ≠ T1, akkor ez megváltoztatja a folyadékfürdő hőmérsékletét.
(Elvileg igen, a hőkapacitások közti óriási különbség miatt azonban a gáz hőmérsékletváltozása gyakorlatilag mérhetetlenül
kis változást okoz a folyadékfürdő hőmérsékletében). Gay-Lussac nem kapott hőmérsékletváltozást, ezért arra következtetett:
T2 = T1
Ma már tudjuk, hogy az állítás helyes, de mint a fenti kísérletből levont mérési következtetés nem fogadható el. Gay-Lussac
kísérletének mégis fizikatörténeti jelentősége van. Az a felismerés, hogy az ideális gáz szabad tágulásakor hőmérséklete nem
változik meg, mind az energiafogalom kialakulásában, mind az ideális gáz kinetikus modelljének felállításában fontos szerephez
jutott.
Idézzük csak fel Robert Mayer nevezetes gondolatkísérletét! A hő és a munka közti kapcsolat felismeréséhez vezető egyik fontos
gondolata az volt hogy amennyivel több hő kell az ideális gáz 1°-kal való felmelegítéséhez állandó nyomáson mint állandó
térfogaton, annyi munkát végez a gáz az izobár tágulás közben. Dehát ebben a kétféle folyamatban azonos kezdőállapot esetén
a gáz két különböző végállapotba jut! A következtetés pedig csak akkor jogos, ha ugyanabból a kezdőpontból ugyanabba a
végállapotba jut mindkét esetben a gáz. Ehhez a kiegészítéshez volt szüksége Robert Mayernek Gay-Lussac kísérletére
(2. ábra).
Az 1 állapotból kétféle módon visszük át a gázt a 2 állapotba:
- munkavégzés nélkül, csak hőközléssel. Ez két lépésben hajtjuk végre: először állandó térfogaton felmelegítjük
ΔT-vel, majd engedjük szabadon tágulni (miközben hőmérséklete a Gay-Lussac kísérlet tanulsága alapján
állandó marad);
- munkavégzés közben, izobár módon, a szükséges hő befektetése mellett.
A hőközlések közti különbség:
Qp - QV, = CpΔT - CVΔT
2. ábra
A végzett munka:
W = pΔV = RΔT mert a folyamat izobár.
Így jutunk az azóta már Robert Mayerről elnevezett összefüggéshez:
Cp - CV = R.
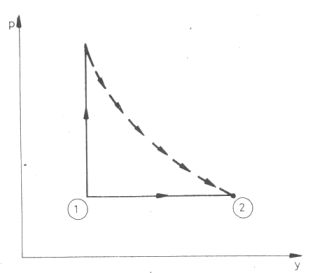
Semmi kétség: a Gay-Lussac kísérlet irreverzibilis folyamat, (ezért is ábrázoltuk a
folyamatot a pV diagramon szaggatott vonallal). A kezdő és végállapot azonban egyensúlyi,
ezért kiszámítható az entrópianövekedés. Ez ugyanannyi, mintha például hőtartályba ágyazva,
izotermikusan vinnénk az ideális gázt a megadott kezdőállapotból a megadott végállapotba, tehát
|
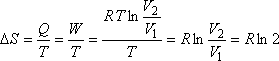 |
|
|
(ha V2 = 2V1) |
|
Csakhogy hőtartályba ágyazva ugyanennyivel csökken a hőtartály entrópiája, s így a
{hőtartály + gáz}
összetett rendszer entrópiája nem változik, ahogyan azt egyensúlyi folyamatok során el is várjuk. Ebben
az egyensúlyi folyamatban a gáz és a hőtartály entrópiaváltozása "kompenzálja egymást" (Clausius
megfogalmazása), míg az irreverzibilis szabad tágulás során a gáz entrópianövekedését nem kompenzálja
semmi a környezetben.
A gáz entrópiája mindkét esetben ugyanannyit változik, mivel a gáz ugyanabból a kezdőállapotból
ugyanabba a végállapotba jut, s az entrópia - állapotjelző.
Fizikatörténeti háttér
A Gay-Lussac-kísérlettel nem az a baj, hogy irreverzibilis, hanem az, hogy a mérési pontosság nem
volt megfelelő.
Ezen igyekezett javítani Joule. Robert Mayer 1845-ben adta ki német nyelven azt a munkáját, amelyben
fent ismertetett gondolatmenetét publikálta s amelyben Gay-Lussac 1807-es kísérletére támaszkodott.
Joule 1843-ban végezte el Gay-Lussac kísérletének módosított változatát a következő módon.
A két azonos térfogatú edényt fémből készítette el, és külön-külön kaloriméterekbe helyezte el őket.
A kaloriméterek térfogata csak kevéssel volt nagyobb, mint a fémedényeké, így viszonylag kis
kapacitású volt a folyadék, ami a fémedényeket a kaloriméterekben körülvette. Ő is a folyadékok
hőmérsékletváltozását mérte, s ebből következtetett a gáz hőmérsékletváltozására.
A kiindulási állapotban az egyik edényt vákuumra szivattyúzta le, a másik edénybe pedig annyi
levegőt pumpált, hogy nyomása 22 atmoszféra lett. Ezután kinyitotta a csapot, s a következő
hőmérsékletváltozásokat tapasztalta: azon az oldalon, ahol eredetileg a 22 atmoszférás levegő volt,
a hőmérséklet 2,36 F°-kal lett kisebb, a másik oldalon a hőmérséklet 2,38 F°-kal lett
magasabb. [5]
Mivel a két kaloriméter hőkapacitása egyenlő volt, ez azt jelenti, hogy ha a mechanikai egyensúly
után sokkal lassabban beáll majd a termikus egyensúly is a két edényben levő gáz között, akkor
a véghőmérséklet a mérési pontosságon belül meg kell hogy egyezzék a kiindulási hőmérséklettel.
Joule körültekintő kísérletező ügyessége tette lehetővé, hogy a Gay-Lussac kísérlet visszanyerte
eredeti bizonyító erejét. Már csak egyedül Joule nem volt megelégedve a mérési pontossággal. Az
az igazság, hogy Joule, amikor eredeti formájában - tehát egyetlen közös kaloriméterben - ismételte
meg Gay-Lussac kísérletét, mégiscsak kapott valamennyi hőmérsékletváltozást, de ez a mérési hiba
határát súrolta.
Azóta, hogy 1807-ben Gay-Lussac megfogalmazta nevezetes tételeit a gázok egyenlő viselkedéséről
a kísérleti módszerek finomodásával egyre több kételkedő kutató lépett fel Gay-Lussac kijelentését
cáfoló mérési adatokkal. A Francia Akadémia már 1811-ben pályázatot hirdetett a minél pontosabb
kísérleti vizsgálatokra. Joule-ban két nézet viaskodott egymással. Egyik a gázok egyforma viselkedését
értelmezni tudó molekuláris elmélet kidolgozásának reménye, amely szerint CV
állandó. Másik a saját és mások kísérletei alapján felmerülő kétely CV
állandóságában. Hogyan növelhető a fajhőmérések pontossága?
Joule 1848-ban, Manchesterben egy tudományos felolvasóülésen kifejti azt a véleményét, hogy
"azok a kutatások, melyeket Victor Regneault a francia kormány megbízásából vállalt a testek
hőkapacitásának fontos problémáját is felölelik, így rövid időn belül új meghatározást várhatunk a
gázok fajhőjére. Ezt a meghatározást ugyanaz a pontosság fogja jellemezni, amely teljesen
megérdemelten szerzett dicsőséget ennek a híres kutatónak." [6]
Ennél a híres kutatónál dolgozott néhány hónapig 1845-ben, miután Cambridge-ben matematikából és
fizikából diplomázott - William Thomson. A Regnault által követelt nagy mérési pontosság indította
Thomsont azokra a vizsgálatokra, amelyek az abszolút hőmérsékleti skála felállításához vezettek.
Megszületett a tökéletes gáz absztrakciója, melyet a valódi gázok csak több-kevesebb pontossággal
közelítenek. Itt Párizsban ásta ki a jótékony(?) feledés süllyesztőjéből a 21 éves Thomson az akkor
már 13 éve halott Carnot-nak 1824-ben (Thomson születésének évében) publikált dolgozatát, és
hazatérve Glasgowba 1848-ban közölte a termodinamikai hatásfokra alapozott abszolút hőmérsékleti
skálát.
Glasgow és Manchester mintegy 300 km-re vannak egymástól. Thomson és Joule szelleme számára
ennél nagyobb távolság sem lett volna akadály barátságuk kialakulásához. Joule hat évvel volt idősebb
Thomsonnál, először 1847-ben találkoztak [7]; lehet, hogy éppen Thomson
hívta fel Joule figyelmét Regnault pontos méréseire.
Ettől kezdve állandó levelezésben álltak egymással. Ha Thomson egyetemi előadásán valami új,
frappáns demonstrációs kísérletet talált ki, azonnal megírta Joule-nak. Ugyanakkor Thomsont, akit
lenyűgözött Fourier munkássága, amely még a hőanyag modelljével írta le matematikailag a hővezetés
elméletét, egyedül Joule kísérleti eredményei győzték meg a hőanyagelmélet tarthatatlanságáról.
[7, 8]
Közös munkájuk legszebb eredménye annak az effektusnak a felfedezése, mellyel ipari méretekben
megindulhatott a gázok cseppfolyósítása, s amely ma valamennyi háztartási hűtőgép működésének
egyik nélkülözhetetlen feltétele, az un. Joule-Thomson effektus.
3. A Joule-Thomson-kísérlet
Vegyünk egy hőszigetelt falú hengert, melyet középen egy porózus anyagból készült fal (pl. vattadugó)
választ két részre (3. ábra).
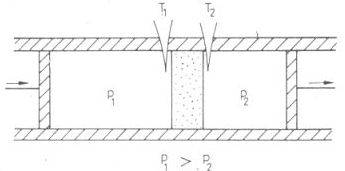
3. ábra
A henger két oldalán 1-1 adiabatikus dugattyút mozgatunk úgy, hogy a porózus fal két oldalán a
nyomás állandóan p1 illetve p2 maradjon, miközben
a porózus falon keresztül a gáz lassan áramlik a nagyobb nyomású oldalról a kisebb nyomású oldalra.
(A porózus fal szerepe az, hogy ugyan megengedi a gáz átáramlását, de nem engedi a gázt felgyorsulni.
Mintegy "lefojtja" a gáz áramlását, ezért a kísérletet gyakran "fojtásos" kísérletnek is nevezik. A fojtás
szerepét egy szűkület is átveheti a csőben, a gyakorlati alkalmazás során gyakran így valósítják meg az
effektust.)
A porózus fal két oldalán termoelemekkel mérjük a T1 és
T2 hőmérsékleteket.
A kísérlet irreverzibilis, bár a gáz kezdő és végállapota (amikor csak a baloldalon illetve
csak a jobboldalon helyezkedik el a kiszemelt gázmennyiség) egyensúlyi állapot.
Ezt az alábbi ábrákkal lehet szemléltetni (4. ábra).
Állítás:
U1+ p1Vl = U2 +
p2V2
Bizonyítás: Az első főtétel értelmében
U2 - U1= Q + W
Most Q = 0 az adiabatikus szigetelés miatt,
W = p1V1 - p2V2
mert a baloldali dugattyú lenyomásakor p1V1 munkát végzünk, a
jobboldali dugattyú elmozdulása közben pedig p2V2 munkát
nyerünk. A befektetett munka így a kettő különbsége.
Átrendezés után kapjuk a bizonyítandó állítást.
A H = U + pV mennyiséget entalpiának nevezzük, így a Joule-Thomson kísérletben
H1 = H2
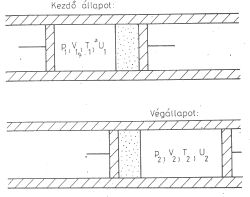
4. ábra
mindig, akármilyen gázzal végezzük is azt el. A gáz új egyensúlyi állapota tehát mindig ugyanolyan
entalpiájú, mint a kiindulási állapot volt.
Ideális gázra pV = RT, tehát U = H - RT, vagyis ez a kísérlet ugyanúgy alkalmas
annak az igazolására, hogy a belső energia csak a hőmérséklet függvénye, mint a Gay-Lussac kísérlet.
Ha pedig T a második főtétellel definiált abszolút hőmérsékletet jelenti, akkor a Gay-Lussac
kísérlethez hasonlóan ez is alkalmas lesz az abszolút hőmérsékleti skála és az ideális gáz hőmérsékleti
skála egyenértékűségének bizonyítására, s közvetve a második főtételbe vetett bizalmunk
megerősítésére.
Csupán azt kell a méréssel ellenőriznünk, igaz-e, hogy T1= T2.
Viszont óriási különbség van a Gay-Lussac kísérlet és a Joule-Thomson kísérlet mérési
megbízhatósága, pontossága között!
Itt közvetlenül a gáz hőmérsékletét mérjük a kis hőkapacitású termoelemekkel, ezért a hőmérsékletmérés
pontossága legalább egy nagyságrenddel nagyobb.
Másrészt a Joule-Thomson kísérletben nem egy meghatározott, korlátozott tömegű gáz
állapotváltozását vagyunk kénytelenek megvizsgálni, mint a Gay-Lussac kísérletben, hanem megfelelő
kompresszort és nyomásszabályozókat csatlakoztatva a henger két oldalára, a nyomásokat
szabályozottan állandó értékeken tudjuk tartani, s így tetszőleges mennyiségű gáz állapotváltozását
tudjuk figyelemmel kísérni. A Joule-Thomson kísérlet során kivárhatjuk a stacionárius (időben állandó)
folyamat kialakulását. Az egyetlen probléma a jó hőszigetelés megvalósítása, az ebből eredő hiba
csökkentésére helyezzük el a termoelemeket a fojtás közelében.
Mivel a Joule-Thomson kísérlet is olyan irreverzibilis folyamat, ahol a kezdő és végállapot
egyensúlyi, ezért itt is kiszámítható az entrópianövekedés, a Gay-Lussac kísérlethez hasonló módon.
Tekintsünk egy egyensúlyi, izentalpikus folyamatot:
TdS = dU + pdV
TdS = d(U + pV) - Vdp
TdS = dH - Vdp
dH = 0, tehát
TdS = -Vdp
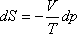
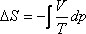 Mivel a Joule-Thomson kísérletben p2 < p1, ezért a
helyettesítő egyensúlyi folyamatban
Mivel a Joule-Thomson kísérletben p2 < p1, ezért a
helyettesítő egyensúlyi folyamatban
dp < 0.
kell, hogy legyen. Tekintettel arra, hogy V > 0, és T > 0, ezért szükségképpen
ΔS > 0.
Egy mól ideális gázra 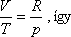
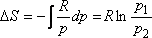
Az egyensúlyi folyamat során ugyanennyivel csökken a folyamatban
résztvevő hőtartályok összentrópiája, viszont az irreverzibilis Joule-Thomson
kísérletben ez a kompenzáló entrópiacsökkenés hiányzik, mivel a gáz
környezettől adiabatikusan el van szigetelve.
Hasonlítsuk össze egy mól ideális gáz entrópiaváltozását a két kísérletben!
Gay-Lussac kísérlet:
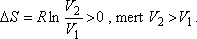
Joule -Thomson-kísérlet:
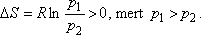
A Gay-Lussac kísérletben V2 / V1 = 2 volt, a Joule-Thomson
kísérletben azonban p1 / p2 = 200 is lehet!
Reális gázok esetén is sokkal nagyobb a tömegegységre
jutó entrópianövekedés a Joule-Thomson kísérletben, mint a Gay-Lussac kísérletben. Ez is oka annak,
hogy a reális gázok esetén mérhető hőmérsékletváltozás sokkal nagyobb a Joule-Thomson kísérletben,
mint a GayLussac kísérletben.
Hőmérsékletváltozások a Joule-Thomson-effektus során
Ha 200 atm-ról 1 atm-ra csökkentjük az eredetileg 52 °C-os levegő nyomását, akkor
hőmérséklete 23 °C-ra csökken. Ha 52 °C-os hélium nyomását csökkentjük 200 atm-ról
1 atm-ra, akkor a hélium 64 °C-ra melegszik fel. Tipikus hűtőközegek az ammónia és a freon 12.
Ha ezek hőmérsékletét akarjuk 30 °C-ról -15 °C-ra csökkenteni a Joule-Thomson effektus
segítségével, akkor az ammónia nyomását például 12 atm-ról 2,4 atm-ra, a freon 12 nyomását pedig
elég 7 atm-ról 1,8 atm-ra csökkenteni.
Általánosan jellemző kísérleti tapasztalat, hogy a kritikus nyomásnál legalább egy nagyságrenddel
nagyobb kezdőnyomásról indulva mind hőmérsékletcsökkenés, mind hőmérsékletemelkedés
lehetséges a Joule-Thomson kísérletben. Ez csak attól függ, hogy milyen a végállapotbeli nyomás.
Kivételt képeznek azonban például az igen magas hőmérsékletű kezdőállapotok, ekkor a
Joule-Thomson effektus mindig a hőmérséklet emelkedéséhez vezet.
Példaképpen nézzük meg a túlhevített vízgőz esetét. Ennek kritikus hőmérséklete
374 °C = 647 K, kritikus nyomása 225 atm. A mérnöki gyakorlatban használatos
vízgőztáblázatokból kiolvasható, hogy ha mondjuk 2 atm-ról 1 atm-ra fojtjuk le a
túlhevített vízgőz nyomását Joule-Thomson-effektus segítségével, akkor 130 °C-ról
indulva 4,5 °C-os hőmérsékletcsökkenést, 165 °C-ról indulva 3 °C-os csökkenést,
500 °C-ról indulva 0,5 °C-os csökkenést kapunk. Ahhoz, hogy a hőmérséklet ne
csökkenjen, hanem emelkedjék, a kiindulási hőmérsékletet jóval 1000 °C fölé kellene emelni.
Olyan magasra amikor viszont a H2O molekulák a nagy sebességű ütközések
következtében részben gerjesztődnének, esetleg ionizálódhatnak is, s ekkor már nem az eredeti gázról
lenne szó. A mérnöki vízgőztáblázatokban nem is találunk 1000 °C-nál nagyobb hőmérséklethez
tartozó adatokat.
A Joule-Thomson-effektus adta hőmérsékletváltozás azonban nem csak a kiindulási hőmérséklettől,
hanem a kiindulási nyomástól is függ. Ha például az 500 °C-os vízgőz nyomását nem 2
atm-ról fojtjuk le 1 atm-ra, hanem 500 atm-ról 499 atm-ra, a hőmérsékletcsökkenés az
előbbi 0,5 °C-nak is csak a fele, 0,25 °C lesz. Ez azt mutatja, hogy elég magas nyomáson
az ilyen kis Δp értékkel való fojtás (ezt nevezik differenciális Joule-Thomson
effektusnak) előbb-utóbb hőmérsékletemelkedéshez vezet.
Persze ha az 500 °C-os, 500 atm nyomású vízgőz nyomását 1 atm-ra fojtjuk, (ez az igazi,
vagy "integrális" Joule-Thomson effektus) akkor hőmérséklete már jelentősen csökken, lehűl
122 °C-ra).
Nemcsak gázzal, hanem folyadékkal is elvégezhetjük a Joule-Thomson kísérletet. A H2O példájánál maradva
megvizsgálhatjuk, hogyan változik a víz hőmérséklete a fojtásos folyamat során. A víz entalpiája elég jó közelítésben csak a
hőmérséklet függvénye, így csak nagyon kis hőmérsékletváltozásokat várhatunk. Induljunk ki itt is az 500 atm nyomású
kezdőállapotból, legyen a kezdőhőmérséklet 273 °C (= 546 K). Ha követjük azt a izentalp görbét, amely ezen a
kezdőállapoton halad át a p; T diagramon, akkor az alábbi p, T értékpárokhoz jutunk:
p; 500 atm 400 atm 300 atm 200 atm 100 atm
T; 273,0 °C 273,2 °C 273,4 °C 273,3 °C 272,8 °C
Ez azt jelenti, hogy ha az 500 atm nyomású 273 °C hőmérsékletű H2O fluidumot 400 atm-ra fojtjuk,
akkor 0,2 °C-kal felmelegszik, ha viszont 100 atm-ra fojtjuk, akkor 0,2 °C-kal lehűl. Ha éppen 125 atm-ra fojtanánk,
hőmérséklete pontosan ugyanannyi lenne, mint a kiindulási állapotban volt.
Nemcsak ideális gázra jellemző tehát, hogy a Joule-Thomson folyamatban nincs hőmérsékletváltozás.
Minden tiszta anyagra, nem túl kicsi és nem túl nagy
hőmérsékletek esetén akármelyik hőmérséklethez tartozik tetszőlegesen sok olyan (p, p') nyomás értékpár, amelyek
entalpiája egyenlő egymással.
Ekkor a nagyobb nyomásról a megfelelő kisebb nyomásra fojtva az anyagot, hőmérséklete nem változik meg. Az előbb említett
273 °C vizet 500 atm-ról 125 atm-ra, vagy 400 atm-ról 175 atm-ra vagy 350 atm-ról 250 atm-ra fojtva nem lesz
hőmérsékletváltozás. A differenciális Joule-Thomson effektus során ezen a hőmérsékleten kb. 300 atm nyomáson nem lesz
hőmérsékletváltozás.
Joule-Thomson koefficiens
A víz most leírt viselkedése a tiszta anyagokra általában is jellemző, s az alábbi ábrákon foglalható össze:
(5. ábra.).
A p; T koordinátarendszerben az olyan p*, T* állapot, amely esetén a differenciális Joule-Thomson-effektusban
nincs hőmérsékletváltozás, mindig egy izentalp görbének a legnagyobb hőmérsékletű állapota. Látszik, hogy ugyanilyen entalpiájú,
de p*-nál kisebb nyomású állapotok esetén differenciális Joule-Thomson-effektus során a hőmérséklet csökken,
p*-nál nagyobb nyomások esetén nő. Ezért a p*, T* állapotot a megfelelő entalpiához tartozó "inverziós
állapotnak" hívják, itt "fordul meg" (invertálódik) a differenciális Joule-Thomson-effektus kimenetele.
A differenciális Joule-Thomson-effektus eredményének mennyiségi jellemzésére vezették be az ún. JouleThomson-koefficienst
(µJ.T.,) az alábbi definícióval:
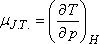
Az egyensúlyi termodinamika alapösszefüggései segítségével µJ.T.-t az anyag más jellemzőivel is
összekapcsolhatjuk:
TdS = dU + pdV
TdS = d(U + pV) - Vdp
TdS = dH -Vdp
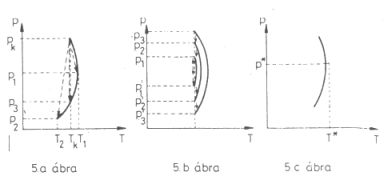
5. ábra. Állandó entalpiájú (izentalp) görbék és fojtásos folyamatok ábrázolása a (p; T) koordinátarendszerben
5. a. ábra. A kezdeti (pk, Tk) állapotból melegebb
(p1, T1) és hidegebb (p2, T2)
állapotba is átmehet az anyag. Ha nyomását éppen p3-ra csökkentjük, hőmérséklete nem változik.
5. b. ábra. Ugyanahhoz a hőmérséklethez tetszőleges sok olyan (pi, pi') nyomás
értékpár tartozik, amelyek esetén nem lesz hőmérsékletváltozás a Joule-Thomson-kísérletben.
5. c. ábra. Egy kiválasztott T* hőmérséklethez tartozó p* nyomás, amely esetén a differenciális
Joule-Thomson effektusban nem lesz hőmérsékletváltozás
Legyenek az extenzív állapotjelzők (S, H, V) p és T függvényei. Ekkor S = S (p,T) teljes
differenciálja:
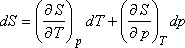
Másrészt
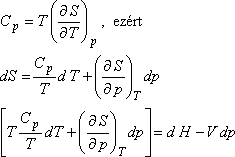
Tekintsük azt a speciális esetet, amikor dH = 0, mivel
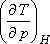 -t
akarjuk meghatározni:
-t
akarjuk meghatározni:
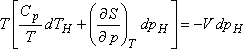
Ebből az egyenletből:
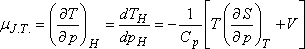
Szemléletesebb kifejezést kapunk, ha felhasználjuk a következő Maxwell relációt:
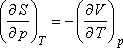
valamint a térfogati hőtágulási együttható alábbi definícióját:
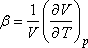
Ezekkel
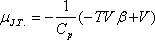
ami így is írható:
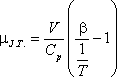
A most levezetett összefüggésünk alapján a Joule-Thomson-koefficiens előjelét
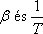 viszonyával tudjuk összekapcsolni.
Az anyagnak azokban az állapotaiban, ahol
viszonyával tudjuk összekapcsolni.
Az anyagnak azokban az állapotaiban, ahol
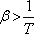 a Joule-Thomson-koefficiens pozitív, tehát a differenciális Joule-Thomson-effektus során az anyag
hőmérséklete csökken.
a Joule-Thomson-koefficiens pozitív, tehát a differenciális Joule-Thomson-effektus során az anyag
hőmérséklete csökken. 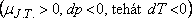 Ha viszont
Ha viszont
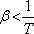 ,
akkor az anyag melegszik. Abban az esetben, amikor
,
akkor az anyag melegszik. Abban az esetben, amikor
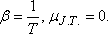 .
Ez éppen az ideális gáz esetén valósul meg, hiszen az ideális gázra
.
Ez éppen az ideális gáz esetén valósul meg, hiszen az ideális gázra
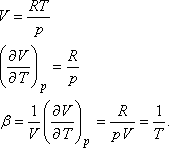
Térjünk vissza a víz példájához. A víz hőtágulási együtthatója
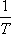 -hez
képest rendkívül kicsi (0 °C és 4 °C között negatív, 4 °C-on zérus, 4 °C és
100 °C között pozitív - 1 atm nyomáson), ezért jó közelítésben a JouleThomson koefficiens
vízre:
-hez
képest rendkívül kicsi (0 °C és 4 °C között negatív, 4 °C-on zérus, 4 °C és
100 °C között pozitív - 1 atm nyomáson), ezért jó közelítésben a JouleThomson koefficiens
vízre:
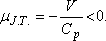
Vagyis a víznek melegednie kell a fojtásos folyamatban. Behelyettesítve a mólnyi mennyiségű vízre
jellemző adatokat:
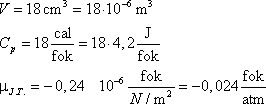
A várható hőmérsékletemelkedés 1000 atm nyomásesés esetén 24 °C. Ez a becslés jól egyezik
a víz-vízgőz táblázatokból kiolvasható értékkel.
Felmerül a kérdés: a szokásos, szobahőmérsékletű víznek nincs inverziós pontja? Miért mindig csak
melegszik a Joule-Thomson-effektus során?
Említettük, hogy az inverziós állapot minden tiszta anyagra így H2O-ra is csak
nem túl kicsi és nem túl nagy hőmérsékleteken léphet fel. Ez a hőmérsékletintervallum nagyon tág, és
általában a hármasponti és a kritikus hőmérséklet közötti értéktől a kritikus (abszolút) hőmérséklet
többszöröséig tart. Vízre az alsó határ 234 °C körül van (kb. 507 K). Ennél alacsonyabb
hőmérsékleteken a folyadék víz csak melegszik a fojtásos folyamatban.
Az inverziós görbe
Az inverziós állapotokat ábrázoló pontok a p;T koordinátarendszerben egy maximummal
rendelkező görbén helyezkednek el, amely valahol a folyadék-gőz fázishatár görbéről indul jóval a
kritikus hőmérséklet után éri el újra a T tengelyt (6. ábra).
Ezt az utóbbi határhőmérsékletet, amely a lehetséges legnagyobb inverziós hőmérséklet nevezik gyakran
egyszerűen inverziós hőmérsékletnek. Fontos jelentése van: ha Joule-Thomson effektussal
hőmérsékletcsökkenést szeretnénk elérni, akkor az anyag kiindulási hőmérsékletének ennél kisebbnek
kell lennie. H2O esetén persze ez egy olyan óriási hőmérséklet, hogy a kérdés feltevése
se indokolt. Az inverziós hőmérséklet levegő esetén még 330 °C, hidrogén és különösen hélium
esetén azonban jóval a szobahőmérséklet alatt van. Hidrogénre -71 °C = 202 K, ami a hidrogén
kritikus hőmérsékletének (33,3 K-nak) még mindig kb. a hatszorosa. Nitrogénre a maximális inverziós
hőmérséklet a kritikus hőmérséklet ötszöröse, héliumra pedig 7,7-szerese (kb. 40 K).
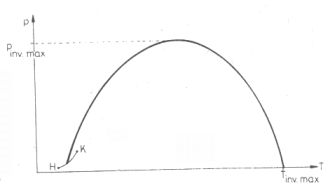
6. ábra. Inverziós görbe a (p; T) koordinátarendszerben.
H = Hármaspont (a gáz, a folyadék és a szilárd fázis közös egyensúlyi állapota),
K = Kritikus pont (a H-t K-val összekötő, ún. folyadék-gáz fázishatárgörbén
vannak a telített gőz állapotok)
Tinv.max. = a legnagyobb inverziós hőmérséklet
pinv.max. = a legnagyobb inverziós nyomás
Az inverziós görbe alatti tartományban µJ.T. > 0, az inverziós görbén
µJ.T. = 0, felette pedig µJ.T. < 0.
A folyadék-gáz fázisgörbe alatt a Joule-Thomson-
koefficiens pozitív, tehát pl. a vízgőzt 234 °C hőmérséklet alatt is lehet hűteni Joule-Thomson-effektussal.
(234 °C-on és 30 atm nyomáson éri el H2O esetén az inverziós görbe a víz-vízgőz
fázishatárgörbét). A 10 atm-ás 190 °C hőmérsékletű vízgőz hőmérséklete 163 °C-ra csökken,
ha fojtással a nyomását 1 atm-ra csökkentjük. A 2 atm-ás 120°°C-os vízgőz hőmérséklete
111 °C-ra csökken, ha 1 atm-ra tágul ki a fojtásos folyamatban (kuktafazék szelepén kiáramló eset),
az 1 atm-ás 100 °C-os vízgőzt 0,01 atm-ra fojtva pedig 93 °C lesz a hőmérséklete.
Ha összehasonlítjuk különböző gázok inverziós hőmérsékleteinek mért értékeit, ez ugyanolyan nagy
változatosságot mutat, mint amilyen változatosak a kritikus hőmérséklet mért értékei. A kettő
hányadosa azonban már sokkal kevésbé változik. Tájékozódásul álljon itt néhányjellemző adat:
[9]
| Tkr | Tinv.max |
Tinv.max/Tkr |
| N2 | 126 K |
621 K | 4,9 |
| levegő | 132 K |
603 K | 4,6 |
| Ar | 151 K |
723 K | 4,8 |
| H2 | 33,3 K |
202 K | 6,1 |
| He | 5,2 K |
40 K | 7,7 |
| pkr | pinv.max |
pinv.max/pkr |
| N2 | 33,5 atm |
400 atm | 12 |
| H2 | 12,8 atm |
106 atm | 13 |
| He | 2,3 atm |
37 atm | 16 |
A fenti táblázatban nemcsak a maximális inverziós hőmérsékletet, hanem az inverziós görbe
maximumához tartozó nyomást is összehasonlítottuk a kritikus állapothoz tartozó megfelelő
értékekkel.
Érdemes ezek után megnézni, hogy a reális gázokra leggyakrabban használt állapotegyenletek -
melyeket molekuláris fizikai meggondolásokból kaphatunk mennyire jól adják ezeket az értékeket. A
legismertebb ilyen állapotegyenlet a van der Waals állapotegyenlet:
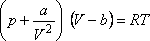
Az ebben szereplő a, b, R paraméterekkel a kritikus térfogat, nyomás és hőmérséklet az
alábbi módon fejezhető ki (l. pl. [10]):
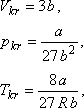
Mint azt az alábbiakban meg fogjuk mutatni, a van der Waals állapotegyenlet valóban egy maximummal rendelkező inverziós
görbét szolgáltat, ahol a maximális inverziós hőmérséklet:
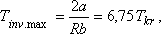
és a maximális inverziós nyomás:
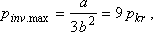
Ezt a maximális értéket az inverziós görbe a
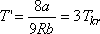
hőmérséklet esetén éri el.
Érdekes még a van der Waals inverziós görbe másik metszéspontja a T tengellyel. Ez nem T" = 0-nál van,
hanem 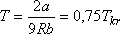 -nál. (Emlékezzünk vissza, hogy H2O esetén a mérési
adatok szerint az inverziós görbe 507 K-nél metszi a folyadék-gőz fázishatárgörbét. Ez a H2O kritikus
hőmérsékletének - 647 K-nek 0,78-szorosa).
Összehasonlítva a van der Waals állapotegyenlet által jósolt
-nál. (Emlékezzünk vissza, hogy H2O esetén a mérési
adatok szerint az inverziós görbe 507 K-nél metszi a folyadék-gőz fázishatárgörbét. Ez a H2O kritikus
hőmérsékletének - 647 K-nek 0,78-szorosa).
Összehasonlítva a van der Waals állapotegyenlet által jósolt
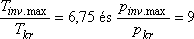
értékeket a fenti táblázat mérési adataiból számolt hányadosok megfelelő értékeivel, azt mondhatjuk, hogy a van der Waals
állapotegyenlet viszonylag jól jelzi az inverziós görbe menetét.
Hélium esetén még jobb egyezést kapunk, ha a reális gázok Dieterici féle állapotegyenletét használjuk. Ez 1 mólra az alábbi
alakú [11, 12]:
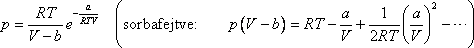
Ha itt is kifejezzük a kritikus adatokat a Dieterici féle a, b állandókkal és R-rel, valamint meghatározzuk
a µJ.T. = 0-hoz tartozó görbét akkor a megfelelő hányadosokra az alábbiakat kapjuk:
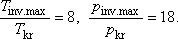
Határozzuk most meg az inverziós görbe egyenletét van der Waals gázra!
Ehhez 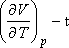 kell meghatároznunk, mivel
µJ.T. = 0,
ha
kell meghatároznunk, mivel
µJ.T. = 0,
ha 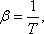 , vagyis
, vagyis
 A van der Waals állapotegyenlet:
A van der Waals állapotegyenlet:
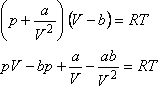
Differenciáljuk ezt parciálisan a hőmérséklet szerint,
állandó nyomás mellett:
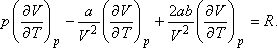
Innen
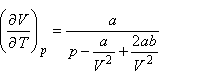
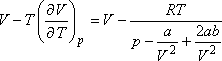
A Joule-Thomson-koefficiens tehát akkor lesz zérus, ha
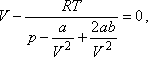
amit átalakítva így is írhatunk:
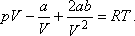
Ez a feltételi egyenlet az állapotegyenlettel együtt az inverziós görbe V paraméteres megadásának tekinthető a p;T
koordinátarendszerben. A két egyenletből
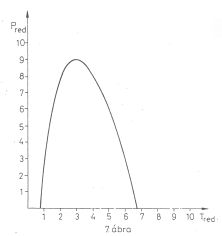
7. ábra A Van der Waals gáz inverziós görbéje;
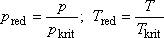
ügyes matematikai átalakítások segítségével V kiküszöbölésével megkaphatjuk az inverziós görbét leíró p(T)
függvény explicit kifejezését is. Ez a következő:
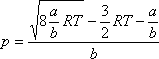
Még áttekinthetőbb és érdekesebb lesz ez a kifejezés, ha az állapotjelzők redukált értékeire írjuk fel. Tekintettel arra, hogy
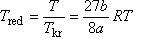
és
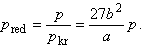
van der Waals gáz esetén, a következőt kapjuk:
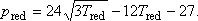
Ennek a függvénynek a zérushelyei:
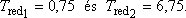
Maximumhelye: Tred.max = 3-nál van, a maximál nyomás redukált értéke itt:
pred.max = 9
.
A van der Waals inverziós görbe menete a 7. ábrán látható.
Gyakorlati alkalmazások
A múlt század utolsó évtizedeiben a gázok cseppfolyósításának nagyszabású programja többek
között. éppen a Joule-Thomson-effektus felfedezése nyomán bontakozhatott ki.
Ugyancsak a Joule-Thomson-effektust használják fel napjakban az ipari és háztartási hűtőgépekben,
valamint a légkondicionáló berendezésekben.
Mindezek tárgyalása azonban már meghaladná e cikk kereteit, a gyakorlati alkalmazások témája
külön tanulmányt érdemel.
IRODALOM
[1] Radnai Gyula: Egyszerű rendszerek állapotegyenletei, ELTE TTK. Szakmódszertani
Közlemények X. 13-37. o. (1977).
[2] Radnai Gyula: Egyszerű rendszerek egyensúlyi állapota és a termodinamikai
állapotfelület, Fizikai Szemle XVIII. 223-237. o. (1978).
[3] Czógler Alajos: A fizika története életrajzokban, Természettudományi Könyvkiadó
Vállalat, Budapest (1882) II. kötet; 97. o.
[4] Gay-Lussac, J. L.: Memoire sur la dilatation des gaz et des vapeurs, Ann. de Chimie,
XLIII. (1802).
[5] Kudrjavcev, P. Sz.: A fizika története, Akadémiai Kiadó, Budapest (1951) 490. o.
[6] Kudrjavcev, P. Sz.: A fizika története, Akadémiai Kiadó, Budapest (1951) 555. o.
[7] Simonyi Károly: A fizika kultúrtörténete, Gondolat Kiadó, Budapest (1978) 314. o.
[8] Mac Donald: Faraday, Maxwell and Kelvin, Doubleday Anchor Books, New York (1964) 114. o.
[9]Zemansky, M. W.: Heat and Thermodynamics,
Mc Graw-Hill Book Co., New York (1968) 340. o.
[10] Budó Ágoston, Pócza Jeno: Kísérleti fizika I. Tankönyvkiadó, Budapest (1962) 456. o.
[11] Parsonage, N. G.: The Gaseous State, Pergamon Press, London (1966) 26. o.
[12] Kubo, R.: Thermodynamics, North-Holland Publ. Co. Amsterdam (1968) 125. 0.